Harnessing biomolecular motion to design novel therapeutics that precisely reprogram protein function
drug discovery
Target insights
Modeling biomolecular motion uncovers novel therapeutic approaches
Considering the dynamic behavior of proteins expands the design space of therapeutics. Here, we simulate the dynamic protein-protein interface to pinpoint optimal strategies for designing molecules that maximize the intended conformational and therapeutic effect.
Generative design
Computational models generate diverse molecular designs aligned with the therapeutic hypothesis
Here, we search the design space for molecular glues that will reprogram protein function by stabilizing the desired protein-protein interface.
Computational assay
Predictive models enable accurate and high-throughput evaluation of therapeutic potency of molecular designs
Computational models forecast not only the affinities of the designed molecules for the target proteins but also their effects on functions. Here, we interrogate the efficacy of therapeutic degraders in tagging a protein for degradation. The therapeutic degraders induce transfer of ubiquitin to the surface lysines on the target protein.
Accelerating discovery
Our binding free energy calculations enabled discovery of twice as many bioactive molecules from half as many synthesized.
Expanding the design space
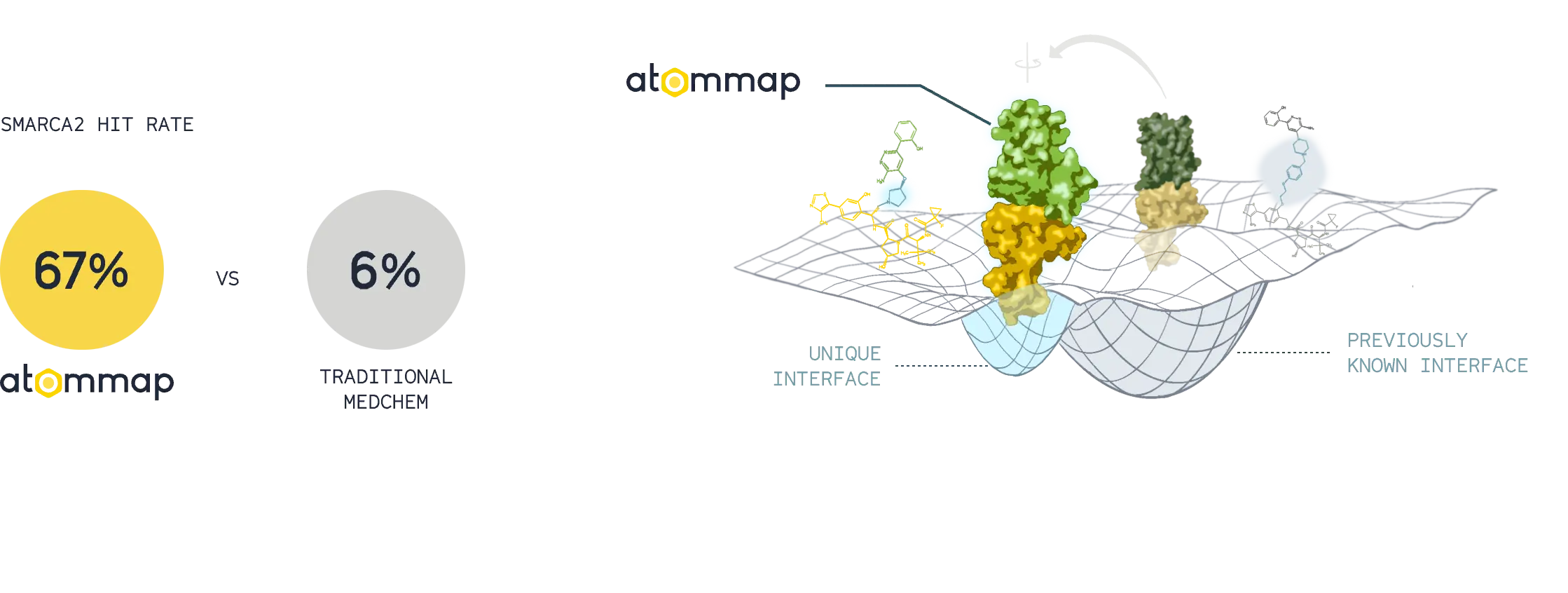
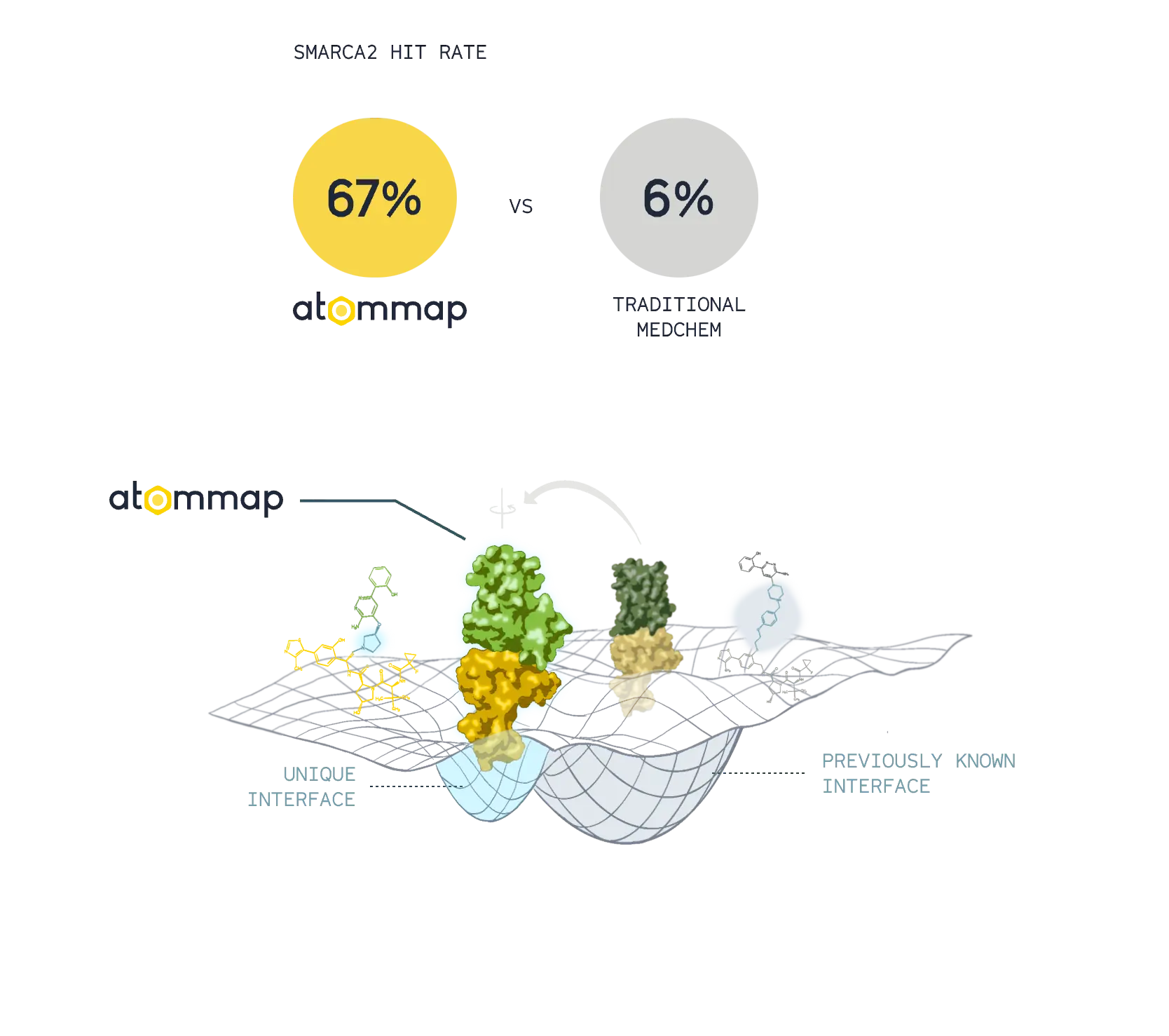
Accurate models of protein-protein interface and computational assays resulted in superior rates of successful designs and unique degrader molecules.
First-in-class molecules
Our leadership team has been instrumental in many of the most impactful developments in computational drug discovery:
- The most widely used software for predicting protein-ligand affinities (FEP+)
- The fastest molecular dynamics simulation software (Desmond)
- A proprietary toolkit for rational design of degraders and molecular glues
- A computational molecular design platform that produced drug molecules advanced into clinic (STING agonist)

Huafeng Xu, PhD
Founder and CEO
Chief Technology Officer of Silicon Therapeutics and Roivant Discovery
Led platform development at D. E. Shaw Research as the founding developer of the fastest molecular dynamics simulation code, DESMOND, and the most widely used binding free energy prediction software, FEP+. Ph.D. from Columbia University

Jesus Izaguirre, PhD
Co-founder and Chief Computational Scientist
VP of advanced simulations in Silicon Therapeutics and Roivant Discovery leading platform development of targeted protein degradation
Scientist at D. E. Shaw Research. Former professor of CS and Engineering and co-director of Computer-Aided Drug Design Core at the University of Notre Dame. Ph.D. from University of Illinois at Urbana-Champaign

Yujie Wu, PhD
Co-founder and Chief Technology Officer
Director of Free Energy Methods at Roivant Discovery
15 years at Schrodinger where he led FEP+ development and engineering teams, and contributed to other molecule dynamics based products. Ph.D. from University of Utah

Cheryl Koh, PhD
Head of Discovery Biology
Scientific leader and key contributor in drug discovery programs at AstraZeneca and DeepCure
Core team member for development of first clinical candidate (systemic STING agonist) at Silicon Therapeutics. Ph.D. from Johns Hopkins School of Medicine

Mandana Honu, PhD
Head of Business Development
Chief Scientific Officer and Head of Business Development of Kaleidoscope
Spent 16 years in cancer research before shifting focus to company building, including key roles in therapeutics and platform investing, new company creation, and corporate BD at Resilience. Led product development and sales at Kaleidoscope. Ph.D. from Duke University

Discovery
We deliver promising molecules, development candidates, virtual screening solutions, and generative designs based on unique target insights.
- Finding initial hits
- Scaffold hopping (structural variations)
- Hit-to-lead development
- Lead compound optimization
- Selectivity enhancement
- Target specificity
- Molecular glue
- Chemically induced proximity
Computational modeling
We deliver accurate models and predictions.
- Highly accurate structural and dynamic models
- Druggable pocket identification
- Binding affinity prediction
- Customized functional potency predictions
Out-licensing Atommap molecules
We collaborate with partners to co-develop our drug candidates.
- The only mutant-specific degraders against a mutant oncogenic protein
- Tackling a clear unmet medical need